



2024年11月14日公開
担当:杉江淳 先生
所属:脳病態解析分野 杉江研究室
近年、次世代シーケンサーの普及と技術の進歩により、ゲノム配列を安価かつ迅速に決定することが可能になってきています。その結果、希少疾患や未診断疾患を含む多くの患者のゲノム配列を解析し、診断を確定することが現実のものとなりました。この診断方法は、患者のプライバシーを守るための個人情報保護法、遺伝情報の適切な利用を定めたガイドライン、医療や研究における倫理的な指針の整備に伴い、今後さらに一般的になると考えられます。
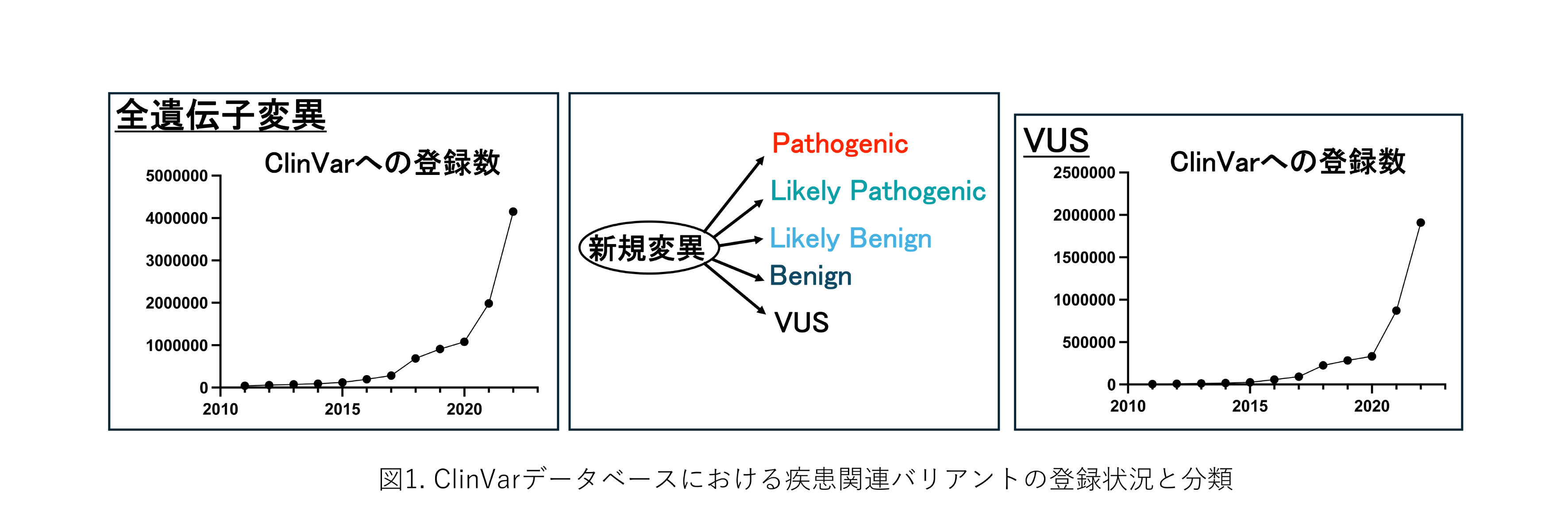
しかし、現在のゲノム解析技術やバイオインフォマティクス技術を駆使しても、病的意義が不明な遺伝子変異候補が年々蓄積されています。実際、ヒトの疾患関連バリアントが登録されているClinVarデータベース[1]では、年々バリアントの登録数が増加しています(図1)。これらのバリアントは、Pathogenic(病原性あり)、Likely pathogenic(病原性の可能性あり)、Likely benign(良性の可能性あり)、Benign(良性)、Variant of Uncertain Significance (VUS)に分類されます。VUSは遺伝子変異が特定されているものの、その変異が疾患に関連しているかどうかがまだ解明されていない状態を指します。ClinVarに登録されるバリアント数の増加に伴い、VUSの数も自然に増加しています。これらのVUSをPathogenicかBenignかに分類するための解析は必要不可欠ですが、病的意義が高いと見込まれない限り、解析にかかる時間やコストが問題となります。現時点では、安価かつ簡便にVUSの意義を評価できるシステムが存在しないため、この状況はジレンマを生んでいます。本コラムでは、VUS解析に関するこれまでの取り組みとその展望について紹介します。
希少疾患とは、一般的な病気と比べて患者数が非常に少ない疾患を指します。世界中では6,000を超える希少疾患が確認されており、これらすべてを合わせると約3億人の患者が存在すると推定されています[2]。希少疾患の定義は国によって異なり、日本では患者数が5万人未満であることや、医療上の必要性が高いことなどの基準が設けられています[3]。希少疾患の多くは遺伝性ですが、ウイルスや細菌感染、環境要因によるケースも見られます[4]。これらの疾患は症状や影響が多様で、同じ疾患でも患者によって症状の現れ方が異なることがあり、診断や治療が難しくなる一因となっています。
希少疾患や未診断疾患に関連する問題は多岐にわたり、患者やその家族に深刻な影響を与えます。これらの問題には、診断の遅れや誤診、臨床的な専門知識の不足、確立された治療法がないことが含まれます。未診断疾患の患者は、正しい診断に至るまでに長い時間がかかり、その過程で誤診を受けることも少なくありません。その結果、不適切な治療が行われ、症状が悪化するリスクが生じます。
また、希少疾患に関する専門的な知識が十分に確立されていないため、患者は自身の症状に対して経験豊富で質の高いケアを提供できる医療従事者を見つけることが難しい状況に置かれています。支援団体は大切な存在ですが、患者数が少ないことから、十分な支援体制が整っていないことが多いです。
未診断の疾患においては、適切な医療支援や心理的サポート、情報提供、経済的な支援が求められています。こうした支援が不十分である現状を改善するためには、さらなる研究と支援体制の充実が必要です。そして、未診断の患者を診断するための手段の一つとして、ゲノム解析などの遺伝情報の解読が期待されています。
2. ゲノム解析技術を利用した診断DNAの変異を調べるための技術には、塩基配列を決定する「サンガーシーケンシング」があります。この技術では、DNAの合成を特定の場所で止め、配列を読み取ります。2003年には、この方法で最初のヒトゲノム配列が解明されましたが、膨大なコストと国際協力が必要でした[5]。この後、シーケンシング技術は大きく進化し、次世代シーケンシングの時代が到来します。
サンガーシーケンシングが単一の遺伝子を対象とするのに対し、2000年代後半から始まった次世代シーケンシングでは、全ゲノムのDNA断片を同時に短いリードに分割し、大規模並列で解析することが可能になりました。この進歩により、以前は1つのサンプルで莫大な費用がかかっていたシーケンシングが、より短期間かつ低コストで行えるようになり、DNA解析の新たな時代を開きました。
2001年から2007年まではサンガー法を基にした「第一世代」シーケンシングプラットフォームが主流でしたが、2008年以降には「第二世代」(または「次世代」)シーケンシング技術の普及が始まりました。この技術革新によって、DNA解析のコストが大幅に削減され、より多くの研究や医療分野での活用が可能となっています[6]。
次世代シーケンシング技術が進化する中で、これらの技術が希少・未診断疾患に適用され始めたのは2008年頃からです。最初の試みは2010年に遺伝性疾患であるミラー症候群の4人にエクソームシーケンシングを適用し、原因となる遺伝子を特定するものでした[7]。4人の患者全員に見られる遺伝子変異を探索した結果、単一の候補遺伝子であるDHODHが特定されました。この遺伝子はピリミジンのデノボ生合成経路の主要な酵素をコードしています。エクソームシーケンシングは、希少なメンデル病の基礎となる遺伝子を特定するための強力で効率的な戦略であり、一遺伝子性形質の遺伝子解析を変革する可能性を見出しました。このように、次世代シーケンシング技術の進歩により、以前は不可能だった診断や新しい治療が可能になりましたが、依然として診断がつかないケースも多く、長鎖リードシーケンシングなど新技術が期待されています[8]。
実際にエクソームシーケンシングによって極めて希少な疾患が診断されても、同じ疾患を持つ患者がほとんどいないため、予後に関する情報が限られ、特定の治療法も提供されないことが多くあります。このため、患者や家族は「遺伝子変異が見つかった」という段階に留まり、その後の対応が不透明に感じられることが少なくありません。しかし、遺伝子検査が可能になってから、患者やその家族がどのような気持ちでいるかを追跡した研究があります[9]。この研究は、確定診断や徹底的な検証検査を受けること自体が、患者やその家族にとって精神的な安心感をもたらす価値があることを示しています。治療の選択肢がなくても、また望ましくない新たな情報が得られる場合でも、診断が重要な役割を果たしています。
この分野は、今後さらに研究が必要とされる重要な課題であり、資金提供機関や製薬業界からの支援が不可欠です。しかし、希少疾患は市場規模が限られているため、製薬会社が新たな薬の開発に積極的でないのが現状です。それでも、疾患に関連する遺伝子変異が特定されることは0から1への重要な一歩であり、次の段階に進むための基礎となります。
3. 次世代シーケンサーによる診断の新たな課題の一つ、VUS次世代シーケンサーの発展に伴い、VUS(臨床的意義が不明な変異)もたくさん見つかるという新たな課題が生まれました。VUSは、遺伝子の配列に変異が見つかっても、その変異が病気に関連しているかどうかが不明な状態を指します。人間の遺伝子には、健康を維持するために重要な情報が含まれていますが、時には配列が変わることがあり、このような変異がすべて疾患の原因となるわけではありません。
もし、VUSが1人の患者にしか見つからない場合、その変異が病因であるかどうかを特定するのは難しくなります。こうしたケースでは、複数の患者が同じ変異を持っていることが確認されると、病因である可能性が高まります。類似の遺伝子変異を持つ患者を特定するための「Matchmaker Exchange」といったプラットフォーム[10]の活用が進められており、これによりVUSの解明が進むことが期待されています。
一方で、同じ変異を持つ患者がいない場合は、患者由来のiPS細胞やマウスモデルの作製によって実験的に変異の意義を検証する方法もありますが、時間やコストの制約があるため、確かな証拠が得られない限り、このような解析が進まないケースも多いのが現状です。
4. VUSの解決に向けた取り組み例希少疾患の患者は、通常、長期間にわたり、費用がかかり、精神的な負担の大きい診断の過程を経験します。しかし、この診断過程を短縮する手段の一つとして、比較的シンプルなモデル生物を用いた研究があります。モデル生物を活用することで、遺伝子解析や病態機序の解明が進み、新たな疾患関連遺伝子の同定にもつながります。また、この研究は基礎的な生物学的システムや経路の理解に貢献し、治療法の開発や希少疾患と一般的な疾患メカニズムとの関連性の解明にも役立っています。
ヒトとは異なる姿を持つ生物が、なぜヒト疾患のVUSの研究に使えるのか疑問に思うかもしれませんが、ここで重要となるのがフェノログ(phenolog)という考え方です。フェノログとは、異なる生物種間で共通する表現型の特徴やパターンを指し、進化的に保存された遺伝子や遺伝子ネットワークを通じて類似した形質や機能を示すことを基にしています。ヒトとは異なる姿のモデル生物であっても、共通の遺伝子やネットワークを持っていることで、ヒト疾患の発見やメカニズムの解明に役立つのです。
たとえば、Notchシグナル伝達経路はAdams-Oliver症候群、Alagille症候群、Hajdu-Cheney症候群など、ヒトの疾患に関連しています。この経路はショウジョウバエでも研究されており、Notch経路の異常がハエの体毛や羽の異常として現れることが知られています。これをヒトの疾患モデルとして見立てて解析することで、遺伝子経路の詳細な理解が進みます[11]。
希少疾患や未診断疾患のVUSの病態研究において、モデル生物を使った代表的な例は、アメリカのUDN(Undiagnosed Diseases Network)[12]のMOSC (Model organisms screening Center)[13][14]やカナダのRDMM(Rare Diseases Models and Mechanisms)[15]のようなプロジェクトがあります。
MOSCは、線虫、ショウジョウバエ、ゼブラフィッシュという3つの主要なモデル生物の実験的および遺伝的ツールを活用しています。これらは哺乳類ではありませんが、長年の研究成果により、ヒト生物学への貴重な洞察をもたらし、ノーベル賞の受賞にもつながっています。たとえば、線虫を用いたマイクロRNAの研究(2024年)、RNA干渉(2006年)、アポトーシス(2002年)、ショウジョウバエを用いた概日リズム(2017年)や先天性免疫(2011年)、胚発生(1995年)に関する研究などがその一例です。これらのモデルはヒトや霊長類モデル、哺乳類モデルに比べて実験コストが低いため、特に病的意義が不明なVUSの解析を行う際には効率的で、希少疾患の研究において非常に有用です。
臨床・遺伝学・メタボロミクスの精密検査を行っても診断に至らない場合、UDNの臨床サイトは候補となる遺伝子や変異に関する情報をMOSCに提供します。MOSCでは、MARRVELというバイオインフォマティクスツール[16]を用いてデータベース検索を行い、ヒトの遺伝子・変異およびモデル生物の相同遺伝子に関する既存の情報を集約します。変異が優先度の高い候補と見なされた場合、ゼブラフィッシュコア、ショウジョウバエコア、線虫コアの3つのコアのうち、解析に適していると判断されるコアに分配され、遺伝子および変異の機能を評価するための実験が計画・実施されます。これにより、VUSの解明が進み、病態機序が明らかになることで診断の確定を助けたり、新たな疾患の発見につながったりします。
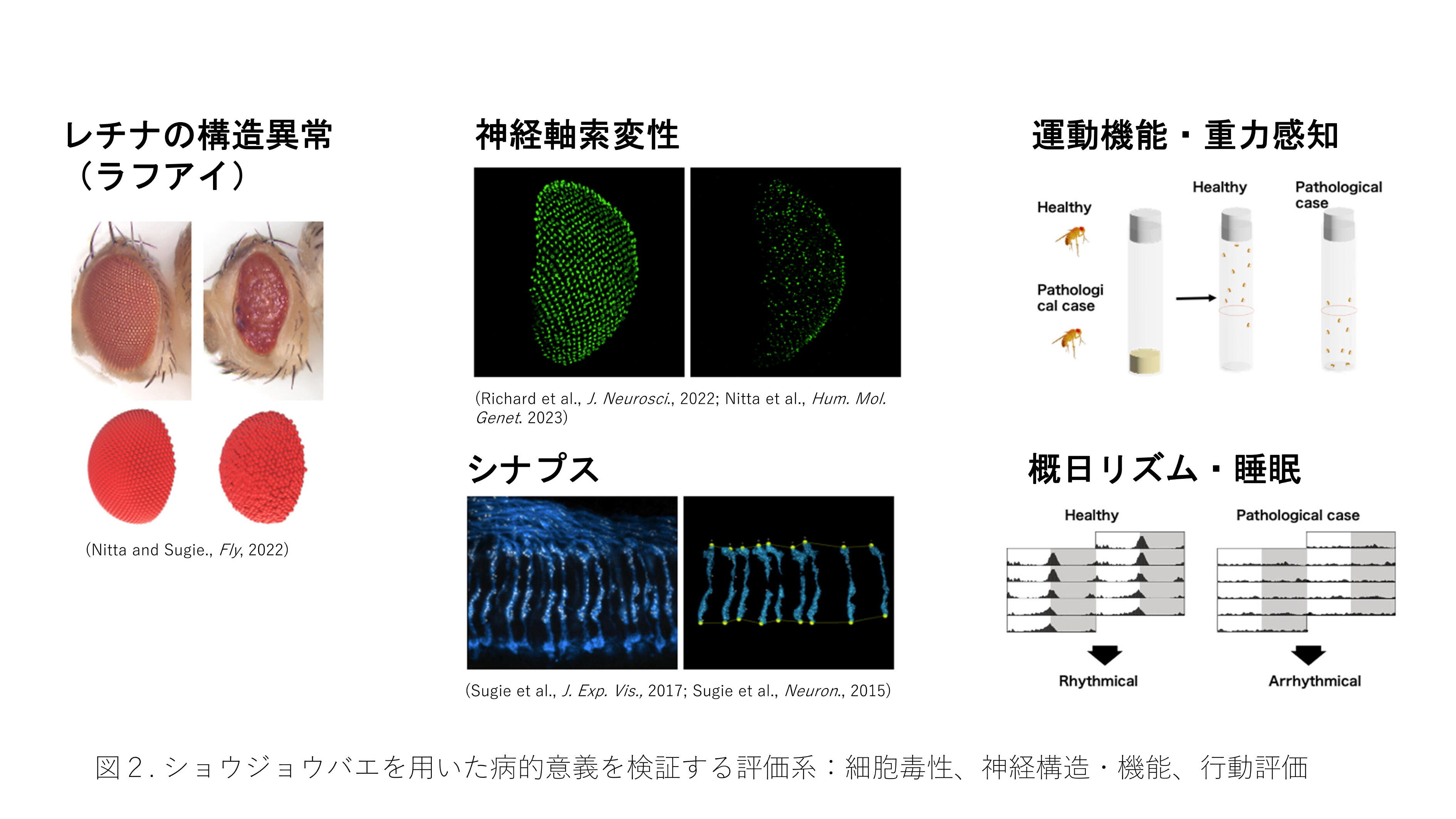
このようなMOSCが中央集権型のシステムである一方で、RDMMは臨床医とモデル生物研究者をマッチングさせる分散型アプローチを採用しています。RDMMでは、カナダの臨床医によってVUSの遺伝子変異が提出され、適切なモデル生物研究者と臨床医がマッチングされてプロジェクトが進行します。このアプローチはカナダで成功を収め、日本(IRUD/J-RDMM)[17]、オーストラリア(AFGN)[18]、ヨーロッパ(Solve-RD)[19]、シンガポール(SingHealth)[20]にもRDMMのようなネットワークが構築されました。日本におけるJapanese Rare Disease Models & Mechanisms Network(J-RDMM)は2017年から2023年度まで活動しており、2024年度からはIRUD(Initiative on Rare and Undiagnosed Diseases、未診断疾患イニシアチブ)[21]の中で、モデル生物解析センターとして引き続きVUSの解消を目指す活動を続けています。私たちもJ-RDMMの研究分担者として、ショウジョウバエを用いたさまざまな評価系を活用し(図2)、これまでにVUSを解消した多くの成果を得ています[22-27]。
MOSCとRDMMには、それぞれ異なる利点と課題があります。MOSCの中央集権型アプローチは、パイプラインの標準化が進んでおり、科学者と臨床医の間で信頼関係を構築しやすいという特徴があります。しかし、比較的少人数でプロジェクトを進行するため、マンパワーの限界からすべての疾患をカバーすることは難しいという課題があります。一方、RDMMの分散型アプローチは、より広範な臨床医と科学者を関与させることができますが、遺伝子ごとに提供される資金やプロジェクト期間が限られていることが問題となります。また、研究のモチベーションや目的が研究者個人に依存するため、すべての解析対象遺伝子において同等の解析を行うことが難しい場合があります。さらに、RDMMシステムは、生物学的な知識が利用可能な変異や遺伝子を研究するには非常に効果的ですが、既存のモデル生物でin vivo研究が行われていない遺伝子は、専門知識を持つ研究者が不足しているため、研究対象として取り組まれない傾向があります。
中央集権型と分散型のアプローチを融合し、中央集権型による効率的な資金提供や遺伝学的ツールセットを構築し、それを分散型のシステムを通じて多くの研究者に提供できれば、さらなる成果が期待できると考えられます。モデル生物を用いた研究は、VUSの病的意義や疾患メカニズムの解明に役立ちますが、それだけでは患者の病気を治療するには不十分です。しかし、希少疾患の患者が多くの場合、治療薬を持たない現状において、治療開発の方向性を探ることは非常に重要です。
VUSが解決しても治療法がない場合、将来的には、アンチセンス核酸医薬や遺伝子治療、ゲノム編集治療などが一般的な選択肢となる可能性があります。
アンチセンス核酸医薬は、特定の遺伝子の働きを抑制することで、病気を引き起こす異常なタンパク質が体内で作られないようにする治療法です。私たちの体内では、遺伝子がさまざまなタンパク質を作り出していますが、その中には病気の原因となる異常なタンパク質も含まれます。アンチセンス核酸医薬治療では、異常なタンパク質の生成を抑えることで、病気の進行を防ぐことが目指されています。VUS変異が機能獲得型毒性を持っている場合効果的です。
アンチセンス核酸医薬は短いDNAやRNAの鎖で構成されており、特定の遺伝子のメッセンジャーRNA(mRNA)に結合する働きを持ちます。mRNAは遺伝子情報に基づいてタンパク質を作る「設計図」のような役割を果たしていますが、アンチセンス核酸医薬がこのmRNAに結合すると、設計図が分解されるか機能しなくなり、結果として異常なタンパク質の生成が止まります。
アンチセンス核酸医薬にはいくつかのメリットがあります。特に、標的となるmRNAに対して選択的に働きかけるため、副作用を抑えながら病気関連のタンパク質生成を精密に調整できる点が特徴です。また、アンチセンス核酸医薬は多くの疾患に対してカスタマイズ可能で、比較的短期間で開発できるという柔軟性も備えています。一方で、課題も存在します。アンチセンス核酸医薬は体内で分解されやすいため、治療効果を持続させるには定期的な投与が必要です。また、アンチセンス核酸医薬を標的となる細胞に効率よく届けることが難しく、治療の実現には配送方法の工夫も重要です。
ここで、アンチセンス核酸医薬の具体例を紹介します。希少疾患の一つである「バッテン病」の患者に対し、個別に治療薬が開発され、治療が成功した症例が報告されました[28]。この症例では、患者と医療チームが接触してからわずか1年で治療が開始され、オーダーメイド医療の実現が示されました。
対象患者の遺伝子検査により、責任遺伝子であるMFSD8に単一の変異(1102G→Cの「ヘテロ」変異)が見つかりましたが、ヘテロであるにもかかわらず病状が発現していたため、両親のどちらかに新たな遺伝子変異がある可能性が考えられました。両親の全ゲノムシーケンス解析の結果、母親にSVA(トランスポゾン)のインサーションが確認されました。この変異がエクソン6のスプライシングに影響を与え、MFSD8の翻訳が途中で停止する原因となっていることが判明しました。この特定の変異は、この患者にのみ見られるものでした。
この変異に対して、アンチセンス核酸医薬を用いたミススプライシングの是正が検討されました。患者から得た細胞を用いた試験で、スプライシング改善に最も効果的なものを特定し、患者の名前にちなみ「ミラセン(milasen)」と命名されました。治療チームは、患者の生命が危機に瀕している状況を考慮し、ラットでミラセンの安全性を確認した後、FDAの許可を得て患者の脊髄への投与を開始しました。
MRIによる詳細な検査では、治療前後で脳萎縮の進行は変わらなかったものの、臨床症状には改善が見られました。具体的には、コミュニケーションや日常生活のスキル、社会性において向上が見られ、フィーディングチューブが不要になるという大きな変化が報告されました。また、1日に15〜30回(持続時間1〜2分)あったてんかん発作は0〜20回に減少し、発作の持続時間も1分以内に短縮されました。
ミラセンが短期間で開発され治療に至った背景には、ヌシネルセンという先例がありました。2016年にFinkelらが報告したヌシネルセン[29]は、脊髄性筋萎縮症1型(SMA1)に対するアンチセンス核酸医薬であり、遺伝子のスプライシングを調整する役割を持っています。この先例があったため、ミラセンの使用を決断することができたと考えられます。
ヌシネルセンの対象となったのは、SMN1遺伝子の欠損によって引き起こされるSMA1で、この病気は乳児期に発症する進行性の運動ニューロン疾患です。
Finkelらは、SMN1に代わる遺伝子としてSMN2に注目しました。SMN2の完全長タンパク質はSMN1と同等の機能を持つものの、スプライシングの過程で75〜90%がエクソン7を欠損してしまい、その結果、機能的にはSMN1の役割を果たしません。そこで、アンチセンス核酸医薬を設計し、エクソン7がスキップされないようにすることで、完全長のSMN2の生成を増加させ、SMN1の不足を補う戦略が採られました。
遺伝子治療遺伝子治療は、遺伝性疾患や一部の非遺伝性疾患の治療を目的として、患者の細胞に新しい遺伝子を導入する技術です。これにより、欠損している遺伝子の機能を補ったり、病気の原因となる遺伝子の影響を緩和することが可能です。遺伝子は通常、病原性を除去したウイルスベクターによって細胞に運ばれます。
遺伝子治療の大きなメリットとして、病気の根本原因にアプローチできる点があります。異常な遺伝子が原因で発症する疾患に対し、正常な遺伝子を導入することで、長期間の効果が期待できます。特に神経細胞では、AAV(アデノ随伴ウイルス)ベクターが高い神経細胞特異性を持ち、神経系での遺伝子導入に適しています。AAVベクターで導入された遺伝子は、細胞分裂が少ない神経細胞でエピソーム形式として安定的に存在し、一度の治療で長期にわたる効果を発揮することが期待されています。これは、進行性や遅発性の神経疾患の治療にとって重要な特性です。
一方で、遺伝子治療にはいくつかの課題もあります。ウイルスベクターを使用することによる安全性の懸念があり、免疫反応が生じたり、導入遺伝子が予期せぬ場所に挿入されるリスクがあることが指摘されています。ただし、AAVベクターの場合、導入された遺伝子は染色体に組み込まれるのではなく、細胞核内でエピソームとして独立して複製されるため、細胞の遺伝的安定性に与える影響は少なく、安全性が高いとされています。
さらに、遺伝子治療は非常に高コストであるため、治療へのアクセスが限られる可能性があります。また、現状では全ての遺伝病や患者に適用できるわけではないため、対象が限定される場合もあります。
遺伝子治療の具体例として、前述のアンチセンス核酸医薬治療と同様に、SMN1遺伝子の欠損によるSMA1の治療が挙げられます。患者に不足しているSMNプロテインを補うため、アデノ随伴ウイルス(AAV)血清型9を用いて、SMN補完DNAを静脈内に一度だけ投与する試みが行われました[30]。この治療には、15名のSMA1患者が参加し、低用量または高用量のベクターゲノムが投与されました。治療の主目的は安全性の確認であり、副次的には換気支援が必要になるまでの時間や運動機能の変化が評価されました。その結果、15名全員が20か月時点で生存しており、これは過去の生存率8%と比較して大幅な改善を示しました。特に高用量を受けた患者では運動機能が向上し、多くが自力で座ったり食事をしたりできるようになり、一部の患者は歩行も可能になりました。この結果は、SMA1に対する遺伝子治療が生存期間の延長や運動機能の改善において有望であることを示していますが、さらなる安全性と有効性の検証が必要です。なお、この治療法は、一度の静脈注射で済むため、脊髄への継続的な投与が必要なヌシネルセンに比べて、患者の負担が少ないと考えられています。また、AVXS-101という名称で開発され、Onasemnogene Abeparvovecとして販売されているこの治療法は、コスト面でも生涯にわたって投与が必要なヌシネルセンよりも効率的であるとされています[31]。
ゲノム編集ゲノム編集技術は、CRISPR-Cas9などを用いてDNAの特定領域を正確に切断し、遺伝子の欠損を修復したり、特定の遺伝子機能を改変したりすることができる革新的な技術です。これにより、遺伝的疾患の原因となる遺伝子変異を直接修正することが可能になります。この技術の大きな利点は、病気を引き起こす遺伝子変異を一度の治療で修正できるため、永続的な効果が期待できる点です。また、CRISPR-Cas9をはじめとする技術は非常に高い精度で遺伝子を編集できるため、効率的な治療が可能です。
一方で、いくつかの課題もあります。オフターゲット効果と呼ばれる、意図しない遺伝子領域が編集されるリスクがあり、これが安全性への懸念を生んでいます。また、生殖系列細胞への応用においては将来世代に影響を与える可能性があるため、倫理的な議論を引き起こしています。このように、ゲノム編集技術は大きな可能性を秘めていますが、その適用には慎重な検討が求められています。
ゲノム編集による治療は現在多くの研究が進められていますが、このコラムではアンチセンス核酸医薬や遺伝子治療と同様に、SMA1を例に取り上げます。最近の研究では、マウスを用いたSMA1モデルに対してゲノム編集が実施され、治療効果が示されました[32]。SMN1とSMN2は99.9%同じ配列を持っていますが、エクソン7の6番目の塩基が異なり、SMN1ではC、SMN2ではTとなっています。この違いがスプライシングに影響を与え、SMN2からはエクソン7が欠失した不安定なタンパク質が主に作られてしまいます。では、なぜSMN2をターゲットにするのでしょうか。それは、SMN1の変異が治療の対象として難しいためです。多くのケースで、患者はSMN1が完全に欠損していることがわかっています。SMN2のわずかな改変で機能を回復できるため、ゲノム編集の対象として選ばれたのです。この研究ではSMN2のTをCに置き換えるゲノム編集が行われ、その結果、完全長のSMNが発現し、症状の改善が見られました。
終わりにアンチセンス核酸医薬治療、遺伝子治療、ゲノム編集治療を実施するためには、まずVUS(意義不明の変異)を解決することが不可欠です。シーケンサーによる検出から始まり、VUSの解決、病態解明研究、治療法の開発、そして臨床での実用化に至るまで、それぞれのフェーズが重要な役割を担っています。さらに、希少疾患のメカニズムを解明することは、一般的な疾患の病態解明にもつながる可能性があります。これらの取り組みを通じて、新たな治療の扉を開き、より広範な医学研究・基礎研究の進展をもたらすことができると思います。
参考文献
- ClinVar https://www.ncbi.nlm.nih.gov/clinvar/
- Wakap et al., Eur J Hum Genet. 2020 Feb;28(2):165-173. doi: 10.1038/s41431-019-0508-0
- 厚生労働省ウェブサイト https://www.mhlw.go.jp/stf/seisakunitsuite/bunya/0000068484.html
- Chong et al., Am J Hum Genet. 2015 Aug 6;97(2):199-215. doi: 10.1016/j.ajhg.2015.06.009
- Lander et al., Nature. 2001 Feb 15;409(6822):860-921. doi: 10.1038/35057062
- DNA Sequencing Costs: Data https://www.genome.gov/about-genomics/fact-sheets/DNA-Sequencing-Costs-Data
- Ng et al., Nat Genet. 2010 Jan;42(1):30-5. doi: 10.1038/ng.499
- Sullivan et al., Annu Rev Med. 2023 Jan 27:74:489-502.
- Rosenfeld et al., Orphanet J Rare Dis. 2023 Apr 10;18(1):73. doi: 10.1186/s13023-023-02695-5
- Matchmaker Exchange https://www.matchmakerexchange.org/
- Salazar and Yamamoto. Adv Exp Med Biol. 2018:1066:141-185. doi: 10.1007/978-3-319-89512-3_8
- Gahl et al., Mol Genet Metab. 2016 Apr;117(4):393-400. doi: 10.1016/j.ymgme.2016.01.007
- Baldridge et al., Orphanet J Rare Dis. 2021 May 7;16(1):206. doi: 10.1186/s13023-021-01839-9
- MOSC (Model Organisms Screening Center) https://undiagnosed.hms.harvard.edu/research/model-organisms-phase-ii/
- Boycott et al., Am J Hum Genet. 2020 Feb 6;106(2):143-152. doi: 10.1016/j.ajhg.2020.01.009.
- MARRVEL (Model organism Aggregated Resources for Rare Variant ExpLoration) https://marrvel.org/
- RDMM(Rare Diseases Models and Mechanisms)https://j-rdmm.org/indexEn.html
- AFGN(Australian Functional Genomics Network)https://www.functionalgenomics.org.au/
- RDMM-Europe (Rare Diseases Models & Mechanisms - Europe) http://solve-rd.eu/rdmm-europe/
- Singapore Rare Disease Models and Mechanisms (RDMM) Network https://www.singhealth.com.sg/patient-care/specialties-services/genomic-medicine-centre/about-singapore-rare-disease-models-and-mechanisms-network
- Adachi et al., Eur J Hum Genet. 2017 25(9):1025-1028. doi:10.1038/ejhg.2017.106
- Iida et al., Front. Genet. 2024 15:1383176. doi: 10.3389/fgene.2024.1383176 Functional analysis of RRAS2 pathogenic variants with a Noonan-like phenotype.
- Vetro et al., The American Journal of Human Genetics 2023 110(8):1356-1376. doi: 10.1016/j.ajhg.2023.06.008
- Yamada et al., European Journal of Medical Genetics 2023 66(8):104804. doi: 10.1016/j.ejmg.2023.104804
- Itai et al., Scientific Reports, 2023 Volume 13, Article number: 975. A novel NONO variant that causes developmental delay and cardiac phenotypes.
- Nitta et al., Human Molecular Genetics, 2023 Volume 32, Issue 9, Pages 1524-1528. Direct evaluation of neuroaxonal degeneration with the causative genes of neurodegenerative diseases in Drosophila using the automated axon quantification system, MeDUsA.
- Sakamoto et al., Human Molecular Genetics, 2021 Dec 17;31(1):69-81. doi: 10.1093/hmg/ddab224.
- Kim et al., N Engl J Med. 2019 Oct 24;381(17):1644-1652. doi: 10.1056/NEJMoa1813279.
- Finkel et al., Lancet. 2016 Dec 17;388(10063):3017-3026. doi: 10.1016/S0140-6736(16)31408-8.
- Mendell et al., N Engl J Med. 2017 Nov 2;377(18):1713-1722. doi: 10.1056/NEJMoa1706198.
- Malone et al., J Mark Access Health Policy. 2019 May 8;7(1):1601484. doi: 10.1080/20016689.2019.1601484.
- Arbab et al., Science. 2023 Apr 21;380(6642):eadg6518. doi: 10.1126/science.adg6518.
